The NIH Pragmatic Trials Collaboratory is excited to announce the availability of AMA PRA Category .75 Credit™ and JA credit AH for the Pragmatic Clinical Trials Study Design learning pathway through the Duke University Health System’s Department of Clinical Education and Professional Development.
This interactive learning path provides researchers with essential knowledge to choose the most appropriate study design for their pragmatic clinical trial. The self-paced modules include expert video by Liz Turner, cochair of the NIH Collaboratory’s Biostatistics and Study Design Core, reference materials, and knowledge checkpoints.
These learning activities are free and require about 1 hour to complete. To access the learning path, visit the NIH Collaboratory Training Resource page and learn more about the Pathways to Learning options. Simply click “get this course” and then “sign up” to create an account in our learning management system.
Continuing Education Credits
In support of improving patient care, the Duke University Health System’s Department of Clinical Education and Professional Development is accredited by the American Nurses Credentialing Center (ANCC), the Accreditation Council for Pharmacy Education (ACPE), and the Accreditation Council for Continuing Medical Education (ACCME), to provide continuing education for the health care team.
Physician Credit: Duke University Health System Department of Clinical Education and Professional Development designates this Pragmatic Clinical Trial Study Design learning path activity for a maximum of .75 AMA PRA Category .75 Credit™. Physicians should claim only credit commensurate with the extent of their participation in the activity.
Nurse Credit: Duke University Health System Department of Clinical Education and Professional Development designates this Pragmatic Clinical Trial Study Design learning path activity for up to .75 credit hour for nurses. Nurses should claim only credit commensurate with the extent of their participation in this activity.
Jennifer Kawi, PhD, MSN, FNP-BC, CNE, FAAN
Maria C. and Christopher J. Pappas Family Distinguished Chair in Nursing
Lee and Joseph Jamail Distinguished Professor
Department of Research
Cizik School of Nursing at UTHealth Houston
Hulin Wu, PhD, MS
The Betty Wheless Trotter Professor & Chair
Department of Biostatistics & Data Science
School of Public Health at UTHealth Houston
Jane Bolin, PhD, JD, RN
Senior Research Professor
College of Nursing, UNT Health Science Center at Fort Worth
Regents Professor Emeritus
Texas A&M School of Nursing
Keywords
Auricular Point Acupressure, Chronic pain, Pain management, Rural communities.
Key Points
Auricular Point Acupressure (APA) uses small seeds embedded into tape and placed on areas of the ear that correspond to areas of the body that are in pain. Placement decisions come from the literature on (APA) locations. APA has been shown to stimulate the central nervous system in fMRI studies, decreased pro-inflammatory substances (IL-ꞵ, TNFα), and increased anti-inflammatory substances (IL-4, IL-10).
To scale APA care, our self-managed approach for the Personalized Auricular Point Acupressure for Chronic Pain Self-management in Rural Populations UG3 study used an electronic application that was created to provide participants with tutorial videos, answers to frequently asked questions, and a pain tracking system. A previous study using this self-managed approach in urban areas showed improved physical function and lessoned pain intensity.
The current study focused on APA in rural communities that experience challenges with access to pain care, transportation, staff shortages, and technology and internet access. Building trust with rural communities was very important.
After the APA intervention, participants felt a regained control over pain and were satisfied with improved comfort. Challenges presented with difficulty reaching points on the back of the ear and discomfort while sleeping.
A full UH3 Pragmatic Randomized Clinical Trial is now being organized to expand the reach of the study. This 3-arm trial will randomize 693 participants to a control, APA in-person, or APA remote group. This study aims to determine the effectiveness of APA in chronic musculoskeletal pain, assess the cost-effectiveness, and identify predictive factors for APA treatment response.
Discussion Themes
Supporting rural communities in research requires leveraging existing partners while building new relationships, listening to the needs of the community and adjusting study procedures to meet these needs, and working with community advisory boards to disseminate findings.
The control group for this study was a pain education intervention group. Creating a sham control group for this type of study is difficult because there are many APA points in the ear. Attempting to create a sham control group that targets other points in the ear may inadvertently be influencing other APA points.
An economic evaluation from the BackInAction trial found that an enhanced course of acupuncture for older adults with chronic low back pain was cost-saving from both the Medicare and healthcare sector perspectives.
In a previously published report, the BackInAction research team established that acupuncture significantly improved pain and disability in patients aged 65 years and older. The new analysis shows the treatment also provides significant value to the healthcare system.
The cost-effectiveness analysis, led by Patricia Herman of the RAND Corporation, analyzed data for 672 participants across 3 large healthcare systems. The study compared 3 treatment strategies: a standard 12 -week course of acupuncture plus usual medical care; standard acupuncture enhanced with up to 6 maintenance sessions plus usual care; and usual care alone.
The research team found that enhanced acupuncture reduced annual back pain–related healthcare costs by an average of $491 per participant and reduced Medicare-reimbursed costs by $421 per participant compared with usual care alone. The savings were primarily driven by a significant reduction in non-acupuncture healthcare utilization.
BackInAction, an NIH Collaboratory Trial, was led by co–principal investigators Lynn DeBar of the Kaiser Permanente Center for Health Research and Andrea Cook of the Kaiser Permanente Washington Health Research Institute.
Beyond financial savings, participants in the enhanced acupuncture group experienced:
Significant gains in quality-adjusted life-years, a standard measure of health-related quality of life
An 18.5 percentage-point increase in the number of participants achieving a clinically meaningful improvement in their disability scores
While standard acupuncture was slightly more expensive than usual care, the strategy’s incremental cost-effectiveness ratio of approximately $53,000 per quality-adjusted life-year suggests it may be cost-effective from the perspectives of Medicare and the healthcare sector.
The BackInAction team’s findings are particularly relevant in the context of the Medicare program’s decision in 2020 to begin covering acupuncture for chronic low back pain. The study suggests that the current Medicare benefit, which includes maintenance sessions, aligns with the most cost-effective and beneficial care for this population.
By including a variety of healthcare settings and older adults with multiple medical conditions, this pragmatic clinical trial’s results are intended to be highly generalizable and to inform future treatment policies for the millions of older people in the United States who experience chronic pain.
BackInAction was supported within the NIH Pragmatic Trials Collaboratory through the NIH HEAL Initiative by a grant from the National Center for Complementary and Integrative Health. Learn more about BackInAction.
Left to right: Kayla R. Mehl, Stephanie R. Morain, and Jeremy Sugarman
A new article from the NIH Pragmatic Trials Collaboratory’s Ethics and Regulatory Core offers a comprehensive scoping review of published empirical ethics research related to pragmatic clinical trials. These trials, which are designed to assess evidence-based interventions in real-world settings, present a variety of unique ethical and regulatory challenges.
The review analyzed 82 published studies of ethics issues that have been explored in the context of pragmatic clinical trials. The authors, led by Kayla Mehl at Johns Hopkins University, identified 22 distinct ethical themes, with the 5 most prevalent being consent and disclosure, risk assessment, trust and transparency, operational burdens and implementation barriers, and the role of engagement.
Consent and disclosure: Traditional written informed consent is often impractical in pragmatic trials, prompting the exploration of alternative approaches such as opt-out or general notification.
Risk assessment: Pragmatic trials present challenges in risk assessment, particularly regarding how “minimal risk” is defined and communicated, which complicates regulatory determinations and participant protections.
Trust and transparency: Trust-building practices, such as results sharing and transparent data use disclosures, are essential for fostering participant confidence.
Burdens, barriers, and costs: Institutions, investigators, and research teams face a variety of operational and logistical burdens in pragmatic trials, especially when integrating interventions into routine care and navigating ethics concerns around data governance and data sharing.
Engagement: Engagement practices in pragmatic trials have been limited and inconsistent, highlighting “a persistent gap between the ideal of inclusive, sustained collaboration and the realities of constrained resources, power dynamics, and unclear stakeholder roles.”
The authors point out that most empirical ethics research related to pragmatic trials has been concentrated in the United States and other Western countries and is heavily reliant on surveys and hypothetical scenarios. This limits the generalizability and real-world applicability of current findings. The authors advocate for future research that is geographically inclusive and that employs innovative methodologies, including nested empirical studies within ongoing pragmatic trials, to provide richer, context-sensitive insights.
Mehl completed a postdoctoral fellowship in the ethics and regulatory aspects of pragmatic clinical trials at the Berman Institute for Bioethics at Johns Hopkins University. Coauthor Stephanie Morain is a core faculty member at the Berman Institute and an associate professor of health policy and management at the Johns Hopkins Bloomberg School of Public Health. Coauthor Jeremy Sugarman is the Harvey M. Meyerhoff Professor of Bioethics and Medicine, professor of medicine, and professor of health policy and management at Johns Hopkins and the deputy director for medicine of the Berman Institute.
The NIH Pragmatic Trials Collaboratory this week published a new chapter of its Living Textbook of Pragmatic Clinical Trials. The chapter, Decentralized Pragmatic Clinical Trials, covers activities of a pragmatic trial that can occur remotely—at a location separate from an investigator’s location—such as participant engagement, recruitment, consent, study interventions and procedures, collection of patient-reported outcomes, and follow-up.
The chapter describes special considerations for decentralized trials, such as community health considerations and the vigilance needed to assure data quality, particularly as it relates to adherence with the study intervention, outcome ascertainment, and event monitoring.
Denise van Hout, MD, PhD
Postdoctoral Researcher
Julius Center for Health Sciences and Primary Care
University Medical Center Utrecht, the Netherlands
Keywords
Adaptive platform trial, Regulatory efficiency, REMAP-CAP, Study design, Study startup.
Key Points
Randomised Embedded Multifactorial Adaptive Platform trial for Community-Acquired Pneumonia (REMAP-CAP) began in 2016 as a data driven analysis of ethical, administrative, and logistical and ethical (EARL) delays in clinical trials studying respiratory infections. The goal was embedded trials that are flexible, efficient, and agile to provide clinicians with high-quality evidence to make the best treatment decisions.
REMAP-CAP is a global multifactorial adaptive platform trial with a master protocol that can investigate multiple interventions in different treatment domains for a single disease.
Over 8000 patients in REMAP-CAP were randomized to 44 interventions in 16 different treatment domains between January 2019 and June 2023. Patients could be randomized to more than 1 domain resulting in 15656 randomizations. Enrollment increased during the COVID-19 pandemic.
Dr. van Hout believes that to improve clinical trials, we should treat the challenges as a scientific problem and solve them with the same rigor.
Regulatory requirements and informed consent regulations differed among sites causing confusion for the researchers about what documents should be submitted with the contract and protocol in each country. Drug labeling requirements in some countries also slowed protocol approval. EARL processes also slowed trial initiation and patient enrollment.
It was clear that overall enrollment in the UK outpaced the other 257 sites worldwide. The UK had a shorter period of time to a fully signed study contract and protocol approval compared with sites in other countries (5 days in the UK compared with 183 days in non-UK countries). This quicker time to signed contract was accomplished by either accepting the contract as-is or rejecting the contract – without negotiating small details. The UK was also 3 months faster than non-UK countries at enrolling the first patient after study approval (1 month vs 4 months, respectively) leading to more enrollment and more research questions answered.
In January of 2022 the EU centralized regulatory submission to a single portal (CTIS) to ease and speed the process of starting a new trial.
Discussion Themes
Adaptive platform trials were uncommon before the COVID-19 pandemic, but their value became clear during the pandemic. After the pandemic, REMAP-CAP focuses on different treatment domains for pneumonia. Maintaining the infrastructure for an adaptive platform trial is difficult if there is not a clear need such as there was during the COVID-19 pandemic.
Centralizing approval for trials under one government body could speed the approval process for studies. During times of high need, prioritizing one or two good trials over a lot of smaller trials can also help speed the process.
The NIH Pragmatic Trials Collaboratory Implementation Science Core, led by Devon Check and Hayden Bosworth, has developed a new chapter on implementation to assist study teams with the complex process of using and studying implementation strategies to help implement research findings into clinical care. The chapter includes sections on:
Case studies are used to illustrate how pragmatic clinical trials embedded in healthcare systems use implementation frameworks, including examples from RAMP, BEST-ICU, STOP CRC, TSOS, ABATE, STEP-2, and GRACE.
Pragmatic clinical trials are conducted as part of routine healthcare delivery and often compare an intervention to usual care. To do this, researchers must understand, monitor, and document standard care at participating research sites.
At the NIH Pragmatic Trials Collaboratory’s 2025 Annual Steering Committee Meeting, Duke University’s Emily O’Brian asked a panel of trialists about the strategies they used to define and document usual care. The panelists included Rachel Winer, co–principal investigator (PI) of STEP-2;, Richard Platt, co-PI of INSPIRE; and Christine Goertz, co-PI of IMPACt-LBP.
Key Strategies
Establish a community of individuals who are interested in and committed to answering the research question
Develop relationships: visit each site and have monthly coaching calls
Get commitment in advance from sites to hold their practice constant for the duration of the trial
Have sites complete readiness surveys or feasibility assessments that include questions about potential upcoming quality improvement initiatives
Minimize burden on sites as much as possible
The Navigating the Unknown chapter of the Living Textbook includes descriptions of unanticipated challenges that may occur during the years-long course of a study that can have profound effects on usual care, including:
At the NIH Pragmatic Trials Collaboratory’s 2025 Annual Steering Committee Meeting, Angelo Volandes, co–principal investigator (PI) of the ACP PEACE trial, led a panel of investigators who shared key challenges and lessons learned from their recently launched trials. The panelists included Stephanie Fitzpatrick, PI of the MOMs Chat & Care Study, Elizabeth Wick, co-PI of I CAN Do Surgical ACP, and ChenChen Wang and Robert Saper, co-PIs of TAICHIKNEE.
MOMs Chat & Care Study
Goal: To test the effectiveness of Northwell Health’s MOMs navigation program at 2 levels of intensity designed to facilitate timely, appropriate care for high-risk Black and Latina birthing people and reduce risk for severe maternal morbidity
Key Challenge: Low recruitment
Solutions:
Expanded inclusion criteria to include Hispanic/Latina patients, those with lower risk factor scores, and gestational of less than 17 weeks instead of less than 13 weeks
Added manual review of charts to ensure the patients they telephoned for recruitment are pregnant and to determine the number of weeks of pregnancy
Changed recruitment materials and general approach so that empathy is at the forefront
Goal: To identify a systems-based approach to help older adults undergoing major elective surgery engage in advance care planning decisions
Key Challenge: One site will finish 6 months early but will still have access to the platform for research, which provides an opportunity they do not want to waste
Solution: Use the trial infrastructure to pilot test another intervention in the remaining months of trial time. The plan is to develop and test human-in-the-loop patient-facing generative AI to assess the quality of patient–AI interactions and answer additional questions, potentially laying the groundwork for future trials.
Goal: To determine whether remotely delivered tai chi is feasible across the 4 partnering healthcare systems and if tai chi, compared with routine care, will improve physical health (including knee pain and function), mental health, and healthcare utilization
Key Challenge: Long delays at institutional review board due to backlogs
Solution: The PI understood that many organizations had to cut costs and reduce IRB and regulatory staff, which likely drove delays in regulatory approval. When the study leadership interacted with the IRB, they did so in a generous and curious way, asking how they could help. The regulatory staff acknowledged the problem and suggested that, as the trial is federally funded and meets the regulatory criteria to be considered minimal risk, it should take priority.
Focusing on the trial’s intention is the first step in designing a trial that successfully answers its primary research question. For an introduction, read Promoting Both Internal and External Validity: Designing the Trial to Match Its Intention, which describes considerations for choosing a pragmatic or explanatory approach. While both approaches are valuable, their purposes are different and will lead to different design choices. As a result, designs that are more pragmatic will have conclusions and recommendations that are more useful for clinical or policy decision-making, and designs that are more explanatory will be more helpful in expanding scientific knowledge.
Case Study of Trial Design in a Renal Dialysis Setting
This case study illustrates the design of 2 randomized controlled trials that examined the temperature used in hemodialysis. Using the PRECIS-2 framework, we discuss insights about opportunities and constraints that a renal dialysis setting offers and show how the design of the trials aligned with their intention. Trial 1 was more explanatory in intention, to test a hypothesis about a mechanism of action: that personalizing dialysate temperature would reduce adverse changes in cardiac morphology and function compared with not personalizing the dialysate temperature (Odudu et al 2015). Trial 2 (the MyTEMP trial) was more pragmatic in intention, to help choose between 2 treatment policies by answering the question of whether patients in dialysis centres that personalized dialysate temperature would be better off in terms of cardiovascular death or hospitalization than patients in centers that maintained fixed temperatures (MyTEMP Writing Committee 2022).
We begin with some background on the 2 trials and the PRECIS-2 domains that will guide the step-by-step considerations for situating the trials on the pragmatic-explanatory continuum.
Trial 1 Characteristics (Shaded Blue)
Personalized dialysis temperature compared with 37° C (98.6° F)
Unit of randomization: individual patient
Patients in Nottingham, United Kingdom, enrolled from September 2009 through January 2013
1 year of follow-up
73 patients would have approximately 11,000 hemodialysis sessions during the trial
Individual-level consent
Trial-specific data collection
Primary outcome: change in the resting ejection fraction measured by cardiac magnetic resonance imaging (MRI) at 12 months compared with baseline
Cardiac structure, function, and aortic distensibility assessed by cardiac MRI
Trial 2 (MyTEMP) Characteristics (Shaded Red)
Personalized dialysis temperature compared with 36.5° C (97.7° F)
Unit of randomization: centers in Ontario followed from April 2017 to March 2021
Maximum 4 years of follow-up
84 centers, 15,413 patients (approximately 6000 entered at start of the trial, approximately 9400 entered during the trial)
Approximately 4 million hemodialysis sessions during the trial
Altered consent by patient notification via poster and newsletter, allowing opt-out by patients or providers
Baseline and follow-up data from large administrative databases
Primary outcome: a composite of cardiac death or hospital admission with myocardial infarction, stroke, or congestive heart failure
Evaluating the Trials Using PRECIS-2
For study teams, the broad steps of evaluating a trial using the PRECIS-2 wheel include:
Defining the trial’s intention
Aligning the design to the intention
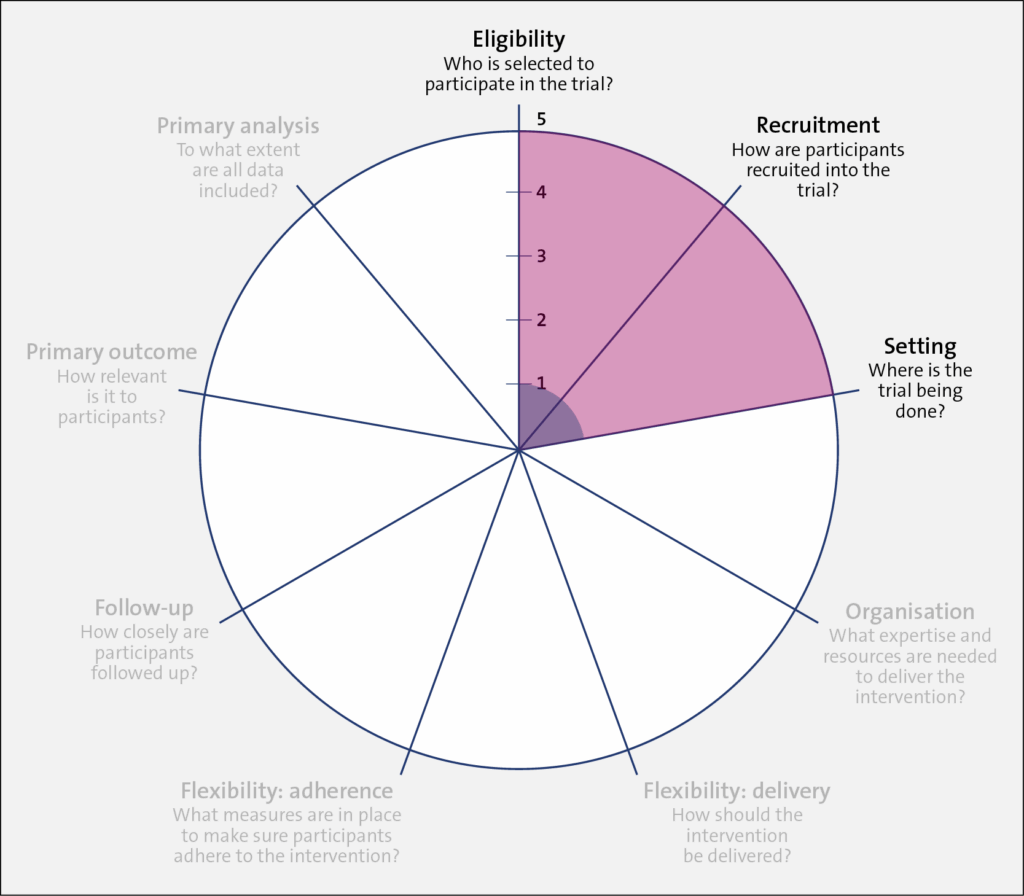
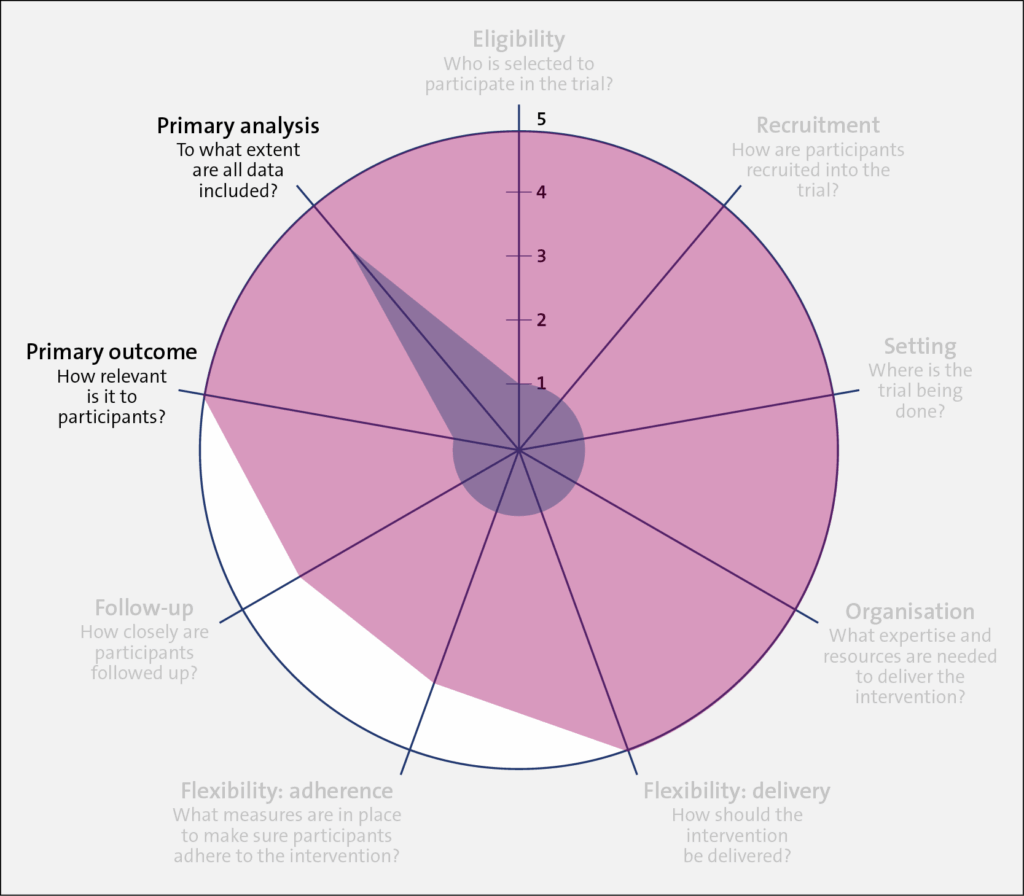
Identifying the trial’s location on the explanatory-pragmatic continuum (from 1 to 5) for each of the 9 domains (the spokes of the wheel)
Once the intention of the trial is set (pragmatic or explanatory), the designer should align their design choices to their chosen intention across all 9 domains of the PRECIS-2, as outlined in the table below. In this brief discussion, we focus on one of these domains, eligibility, with some mention of the primary outcome.
A very explanatory design approach, with many exclusions and a more physiological outcome (morphology) rather than a patient-centered outcome (mortality), is represented closer to 1 on the PRECIS 2 wheel for each of the domains. Such exclusions may include trial participants who are not adherent to the intervention, who do not respond to treatment, who are at low risk for the primary outcome, and children or older patients. The trial also uses diagnostic tests that are not routine in usual care, to assess both patient eligibility and the primary outcome. On the other hand, a very pragmatic design approach with domains that are set to match as closely as possible those in usual care, that includes almost all patients with the diagnosis, that uses usual diagnostic tests to make the diagnosis, and that chooses outcomes that are directly relevant to patients is represented closer to 5 on the PRECIS-2 wheel.
PRECIS-2: Kirsty Loudon et al. BMJ 2015;350:bmj.h2147. Copyright 2015 by British Medical Journal Publishing Group. Used by permission.
The Table below summarizes the PRECIS-2 domains and the considerations for each domain.
Table. PRECIS-2 Domains and Considerations
PRECIS-2 Domain
Considerations
Eligibility
To what extent are the participants in the trial like those who would receive this intervention if it was part of usual care?
Recruitment
How much extra effort is made to recruit participants over and above what would be used in the usual care setting to engage with patients?
Setting
How different are the settings of the trial from the anticipated usual care setting?
Organization
How different are the resources, provider expertise, and the care delivery organization in the trial and those available in the anticipated usual care situation? Are extra resources added?
Flexibility: delivery
How different is trial flexibility of delivery from flexibility anticipated in usual care?
Flexibility: adherence
How different is trial flexibility in monitoring or encouraging adherence from the flexibility anticipated in usual care?
Follow-up
How intrusive is measurement and follow-up of participants in the trial vs anticipated follow-up in usual care?
Primary outcome
To what extent is the trial’s primary outcome directly relevant to participants?
Primary analysis
To what extent are all data included in the analysis of the primary outcome?
Next, we evaluate the characteristics of both trials and describe the considerations behind each of the 9 PRECIS-2 domains.
Domains: Eligibility, Recruitment, and Setting
In terms of the trial population, Trial 1 (shaded in blue) was restrictive in its inclusion criteria, having individuals aged 16 years and older who started hemodialysis at the renal clinic setting within the last 180 days and who had the capacity to provide informed consent. In addition, individuals were excluded if they could not tolerate cardiac MRI, were pregnant or lactating, or were classified with New York Heart Association class IV heart failure.
In comparison, Trial 2 (shaded in red) had less restrictive inclusion criteria, which widened its applicability to most usual care providers, patient populations, and settings. For efficiency (that is, ease of trial conduct) and to match the nature of policy on dialysate temperature, which is set by each dialysis center, rather than for individual patients, randomization was at the clinic (or cluster) level. In this design, the medical director of each clinic agreed for their clinic to be randomized to either trial arm, and all patients were part of the trial. A waiver of individual patient consent was sought and granted.
Having a highly pragmatic approach for Trial 2 allowed for the rapid enrollment of clinics and patients (both patients receiving chronic dialysis at the time of randomization and future patients starting dialysis). However, because patients at low and high risk for experiencing the primary outcome were included, a large sample size was required, as nonselectivity reduces the effect size.
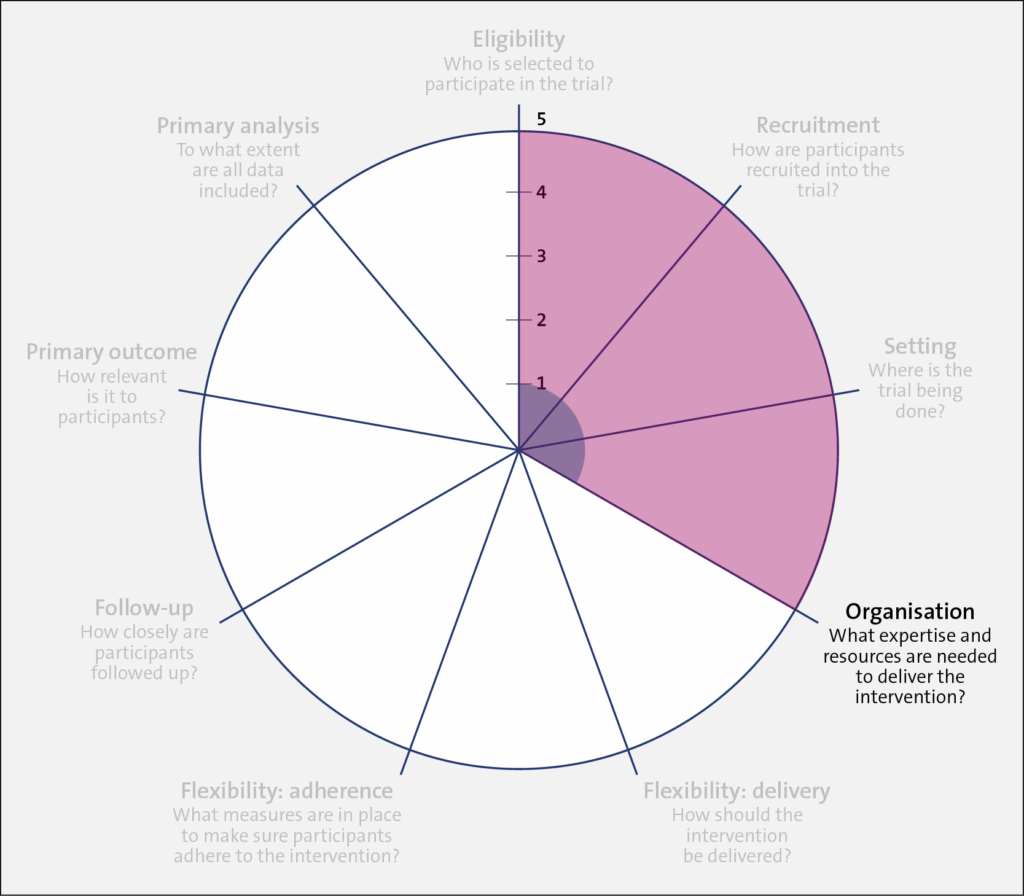
Domain: Organization
Trial 1 (shaded in blue) scored low on the PRECIS-2 wheel because the intervention required a specific type of thermometer, trained research staff, a cardiac MRI, and a dialysis machine with the ability to change the dialysis temperature in increments of 0.1 degrees. In comparison, Trial 2 (shaded in red) scored high on the PRECIS-2 wheel because it required no specific expertise or equipment to implement the assigned treatment protocol.
However, because the Trial 2 protocol became the new clinic policy, there was a need to clearly prescribe and set the dialysate temperature. Also, complex roles and social influences on the process of using a personalized dialysate temperature needed to be assessed.
The flowchart below shows the process of who needed to do what differently, the interrelationships between different roles, and the resulting outcomes. In Trial 2, the leadership at each dialysis clinic changed the local policy to ensure alignment with the assigned temperature protocol. Physicians ordered the assigned temperature protocol for current patients at one time and as new patients received prescriptions for dialysate temperature. Nurses were then trained on the trial protocol and asked to follow the physician's orders to set the dialysate temperature. Nurses were also asked to report any issues or adverse clinical symptoms related to dialysate temperature to the treating physician, who could adjust the dialysate temperature as they deemed appropriate.
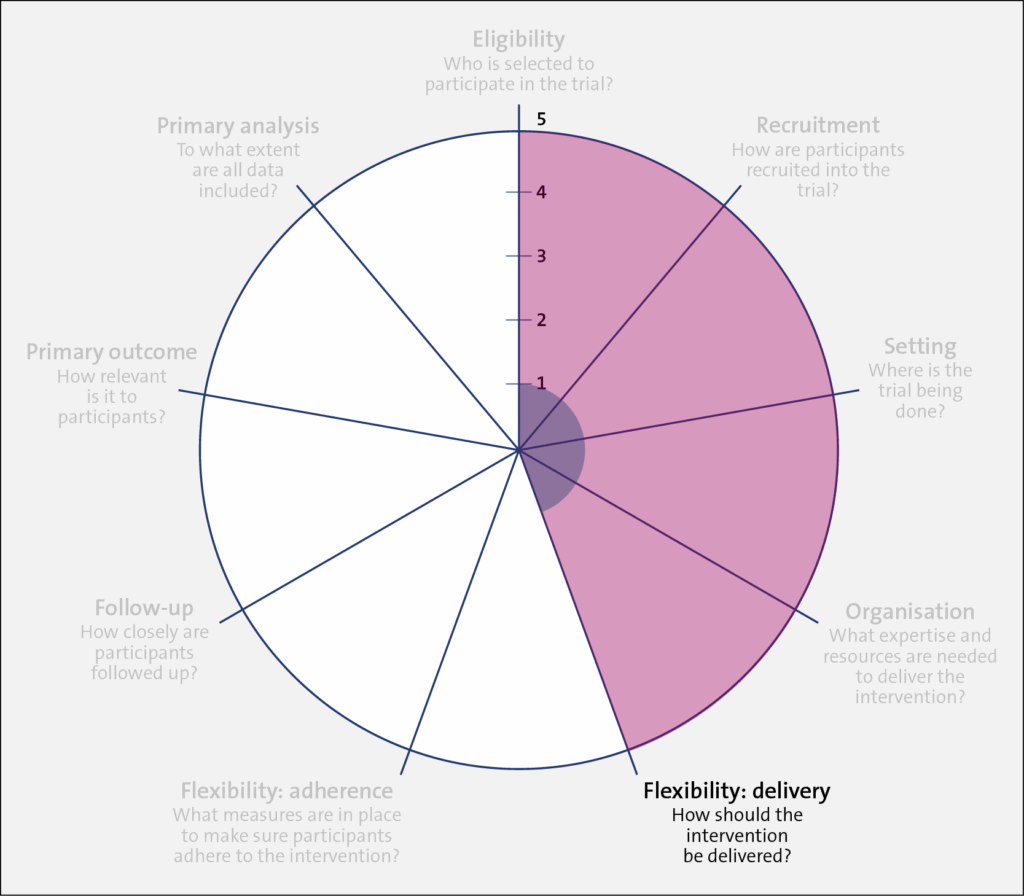
Domain: Flexibility: Delivery
Trial 1 (shaded in blue) required specific dialysis machines, research nurses, and specific thermometers to implement the study intervention. In comparison, Trial 2 retained the dialysis machines in use at each included center and allowed the dialysate temperature in the intervention arm to be set in the range of 0.5 to 0.9 degrees below the patient's body temperature. Also, the entire trial—at the clinic level—was implemented by clinicians and nurses rather than researchers. Little local research infrastructure was required in the recruiting clinics to implement the trial due to this flexibility.
In the control arm of Trial 1 (shaded in red), the dialysate temperature was set to 37° C. In Trial 2, clinics were instructed to set the dialysate temperature to at least 36.5° C, unless the patient or their nephrologist preferred a different temperature.
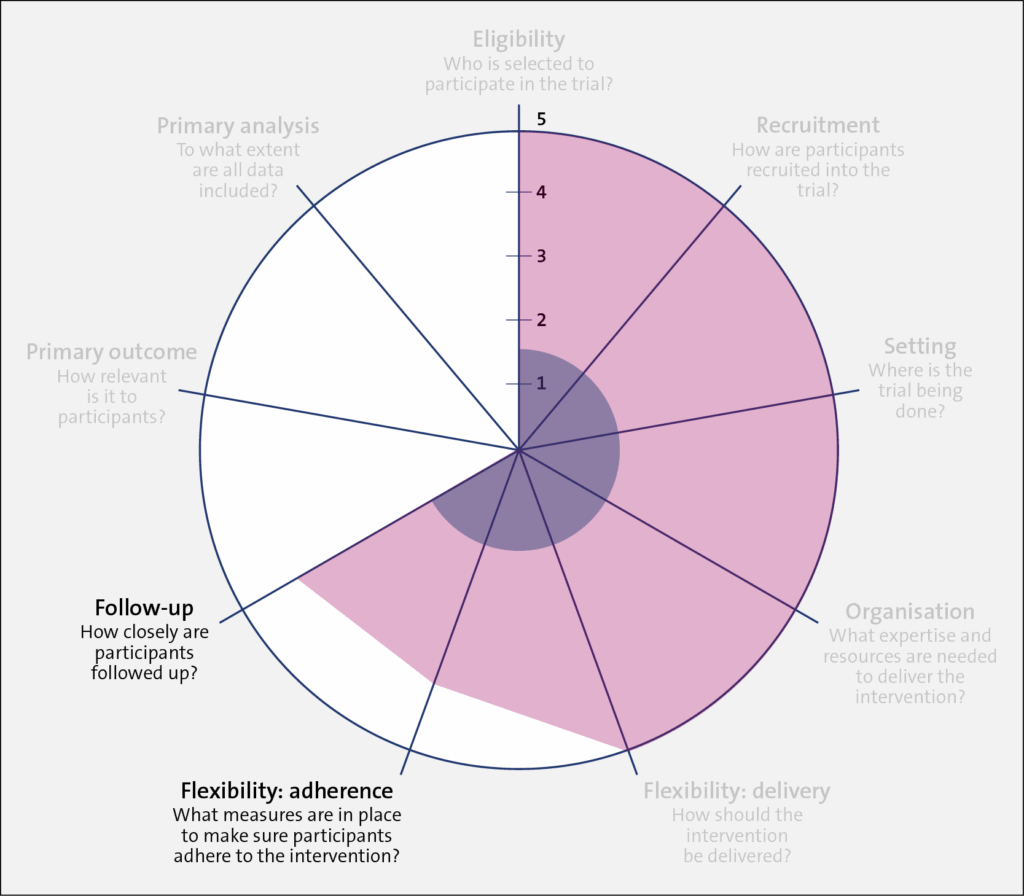
Domains: Flexibility: Adherence and Follow-up
As discussed above, Trial 1 (shaded in blue) employed research staff who closely followed patients for the trial duration. There were regular checkups and feedback sessions between the research staff and participants. In comparison, research staff in Trial 2 (shaded in red) had no contact with participants. However, each month, the clinics were asked to randomly select 15 patients and record the dialysate temperature used for a single session. This allowed the study team to estimate the proportion of patients who were “adhering” to the clinic protocol. It should be noted that the level of adherence for Trial 1 was measured at the participant level, whereas adherence in Trial 2 was measured at the dialysis center. level.
Domains: Primary Outcome and Primary Analysis
Trial 1 (shaded in blue) had a low PRECIS-2 value (1) for the primary outcome because it tested a change in the resting ejection fraction, which is a surrogate outcome. While important, a change in resting ejection fraction is less relevant to patients. In contrast, for Trial 2, the outcome was cardiovascular-related death or hospital admission for adverse cardiac events, which is of greater relevance to patients and their healthcare providers (PRECIS-2 value 5).
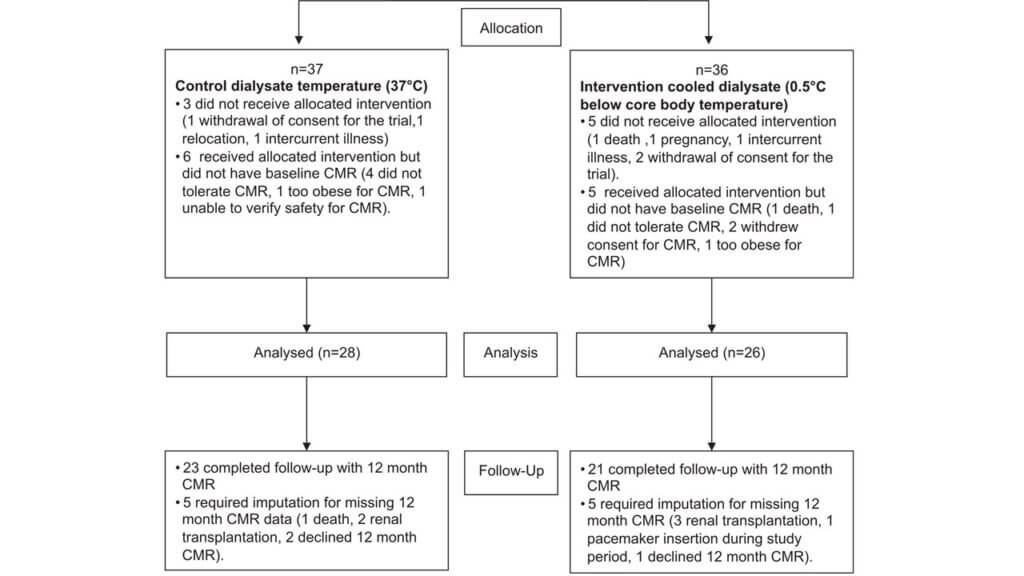
For the analysis domain, Trial 1 used an intention-to-treat approach, which is a highly pragmatic approach. However, nearly 40% of the outcome data was missing due to dropouts or loss to follow-up. From the 73 patients randomized, 37 participants were assigned to the control condition and 36 to the intervention. Of those participants, 28 and 26 were analyzed in the control and intervention arms, respectively. Among those analyzed, data for 5 participants in each arm required multiple imputations for missing outcome data at the 12-month follow-up period.
In contrast, Trial 2 (shaded in red) also used an intention-to-treat approach, but loss to follow-up was expected to be minimal because that typically occurs when study participants leave the (Canadian) province. For example, in Ontario, less than 0.5% the population emigrates from the province each year.
Summary
Pragmatic trials embedded in routine healthcare delivery are increasingly playing a vital role in filling the large gaps in knowledge about caring for patients on hemodialysis. Several fundamental questions in this healthcare setting appear to be particularly suited to pragmatic approaches to trial design. We used Trial 2, the MyTEMP trial, to illustrate a more pragmatic intention, where the authors wanted to test whether the use of a clinic-level protocol of personalized temperature-reduced dialysate results in a different rate of cardiovascular-related deaths or hospitalizations than a standard temperature dialysate. We find it much easier to draw inferences that can be applied to usual care from Trial 2 than we would from Trial 1. The MyTEMP trial was intentionally designed to be highly pragmatic and flexible, which was made possible by:
Frequent and predictable patient encounters
Highly granular and uniform electronic data collection in routine care
Delivery of care by a small number of provider organizations
There is tremendous interest, both nationally and globally, in increasing the momentum for conducting pragmatic trials. Funders of research, including industry sponsors, are increasingly embracing this approach to reduce costs and generate findings that are rapidly translatable to practice.



 The NIH Pragmatic Trials Collaboratory is excited to announce the availability of AMA PRA Category .75 Credit™ and JA credit AH for the Pragmatic Clinical Trials Study Design learning pathway through the Duke University Health System’s Department of Clinical Education and Professional Development.
The NIH Pragmatic Trials Collaboratory is excited to announce the availability of AMA PRA Category .75 Credit™ and JA credit AH for the Pragmatic Clinical Trials Study Design learning pathway through the Duke University Health System’s Department of Clinical Education and Professional Development.
 An economic evaluation from the BackInAction trial found that an enhanced course of acupuncture for older adults with chronic low back pain was cost-saving from both the Medicare and healthcare sector perspectives.
An economic evaluation from the BackInAction trial found that an enhanced course of acupuncture for older adults with chronic low back pain was cost-saving from both the Medicare and healthcare sector perspectives.
 Pragmatic clinical trials are conducted as part of routine healthcare delivery and often compare an intervention to usual care. To do this, researchers must understand, monitor, and document standard care at participating research sites.
Pragmatic clinical trials are conducted as part of routine healthcare delivery and often compare an intervention to usual care. To do this, researchers must understand, monitor, and document standard care at participating research sites. At the NIH Pragmatic Trials Collaboratory’s 2025 Annual Steering Committee Meeting, Angelo Volandes, co–principal investigator (PI) of the
At the NIH Pragmatic Trials Collaboratory’s 2025 Annual Steering Committee Meeting, Angelo Volandes, co–principal investigator (PI) of the