Choosing and Specifying Endpoints and Outcomes
Section 4
Inpatient Endpoints in Pragmatic Clinical Trials
Pragmatic Trial Inpatient Endpoints
In a traditional explanatory trial, data are collected on a case report form (CRF) usually by a study coordinator, but this can be an expensive and inefficient mechanism for gathering data. Explanatory trials also may include outcomes and endpoints that are not typically captured in clinical practice and may not be familiar to health care practitioners. In contrast, embedded pragmatic clinical trials (ePCTs) usually do not rely upon study coordinators for data collection and tend to focus on outcomes that are “directly relevant to participants, funders, communities, and healthcare practitioners (Califf and Sugarman 2015). Pragmatic trials approaches for acquiring patient information may include a variety of data sources (e.g., electronic health records, medical claims and billing data, product and disease registries), patient-reported data, outcomes or surveys, and/or data gathered from wearables or other devices(e.g., mobile phones) (U.S. Food and Drug Administration 2018). These data sources vary in their availability and differ in the type and extent of error and bias they introduce, which can influence study design and results. These differences must be considered when using “real-world” data to ascertain inpatient events.
When an inpatient event is a PCT endpoint, the data necessary to answer a particular research question might vary depending on the level of specificity required. It might be enough to know that the patient was hospitalized in the last six months, or it might be important to know the reason for the hospitalization (e.g., the patient was hospitalized for a heart attack). It may also be necessary to define inpatient events so that they are more meaningful for practitioners and amenable to capture in real-world data sources. In other words, to be more pragmatic in data collection, an investigator may need to be more pragmatic in defining events.
In this section, we will define how inpatient events are classified, describe the different data sources that can be used for inpatient event ascertainment and include information from the literature (where possible) about the reliability of these data sources. For most PCTs, a hybrid approach that uses more than one data source may be the best way to get reliable information as inexpensively as possible (Perkins et al. 2000). We also will provide case studies of pragmatic trials that use inpatient events as endpoints, and propose methods for evaluating the relative accuracy of different inpatient event data collection methods.
Role of inpatient endpoints
In certain therapeutic areas, inpatient events may capture the progression of disease, but this can be an imprecise assessment. Patients can be hospitalized for many reasons. Therefore, depending on what is needed for a trial, just the occurrence of a patient hospitalization may not provide sufficient information.
Current concepts about what constitutes inpatient and outpatient endpoints are grounded in Medicare’s part A and B definitions. Medicare defines hospital status as follows:
- An inpatient stay is when a patient is formally admitted to a hospital. “An inpatient admission is generally appropriate for payment under Medicare Part A when you’re expected to need 2 or more midnights of medically necessary hospital care, but your doctor must order this admission and the hospital must formally admit you for you to become an inpatient (Centers for Medicare and Medicaid Services 2018)”
- An outpatient stay includes emergency department services, observation services, outpatient surgery, lab tests, X-rays, or any other hospital services when the doctor hasn’t written an order to admit a patient to a hospital, even if the patient stays overnight (Centers for Medicare and Medicaid Services 2018).
Other inpatient care, such as stays in rehab and skilled nursing facilities, are defined differently. Further, over the years, as inpatient census acuity has steadily risen in recent years, some care that used to be inpatient, such as diagnostic catheterization and percutaneous coronary intervention, can occur at a hospital, but is considered outpatient (observational) care if the patient stay is not long enough to be considered inpatient. The definition of inpatient care is changing, and there are also regional and health-system related variations in how inpatient care is defined and provided. Therefore, investigators may need to consider not only inpatient stays for outcome ascertainment, but also observation stays and emergency department visits for events that may be equivalent to a hospitalization.
For more, see the article from Medicare: Inpatient or outpatient hospital status affects your costs
How endpoints are classified
Hospitalization endpoints may be classified differently depending on diagnosis. For example, in the 2017 Cardiovascular and Stroke Endpoints for Clinical Trials the definitions for cardiovascular death, myocardial infarction, coronary artery bypass surgery, and stroke do not include the word “hospitalization” (Hicks et al. 2018), although it is likely that the patient was hospitalized for these conditions. The endpoint definitions that explicitly include the term: “hospitalization” are for the conditions that are more challenging to diagnose, i.e., “hospitalization for unstable angina” and “hospitalization for heart failure.” For diseases like cancer, hospitalization is not a common endpoint. Rather, endpoints are based on the progression of the disease. Recommended endpoints for cancer include disease-free survival, time to progression and progression-free survival, and treatment failure (U.S. Department of Health and Human Services 2018). Taking another approach, an FDA perspective on clinical trial endpoints focuses on direct endpoints (survival, symptoms, functioning) and indirect endpoints (biomarkers, walk tests, and tumor size) and does not focus on hospitalization per se (Burke 2012). Nonetheless, many FDA endpoints will occur in an inpatient setting and will require inpatient data for their determination.
In some cases, a hospitalization can be a marker of disease progression, and in other cases, a hospitalization can mean that a patient is getting well enough for treatment. As an example, consider acute heart failure syndrome, where there is a general lack of agreement about appropriate endpoints (Allen et al. 2009). As articulated by Allen et al., there are a number of reasons why hospital stay endpoints are challenging in patients with acute heart failure:
- Preferences regarding staying in hospitals differ across patients
- Regional practice patterns vary
- Patients with a long index hospital stay are less at risk for a repeat hospital stay, because they have less time out of the hospital
- Patients who do not survive are also not at risk for a repeat hospital stay (Allen et al. 2009).
In recent years, systems have been aiming to take care of more heart failure patients outside the hospital setting, which is in line with patient preferences. Thus, meaningful endpoints are aligning with how systems care for patients and what patients prefer for their care.
However, for a pragmatic trial an inpatient hospitalization (acute care, observation, or emergency department) still may be the most appropriate outcome measurement.
Pragmatic Trial Outcomes
There are many factors to consider when choosing what outcomes to measure and how to measure them, including feasibility, interference with practice, and the validity and precision of specific measures (Welsing et al. 2017). Because pragmatic trials focus on problems that are directly relevant to patients and clinician decision-making, the choice regarding outcome should be made with stakeholder preferences and requirements in mind (Welsing et al. 2017). There also are specific considerations related to inpatient events, such as:
- What is the concept of interest?
- What data are needed to answer the research question? What data elements are needed to classify or decide about an event?
- What is routinely measured for the anticipated study population? Are the data commonly documented with consistency during routine care? When and how are the data captured? Does the source provide those data with the right level of specificity and timing?
- What is the quality of the data? Are the measures valid, accurate, and precise?
- What opportunities are available to measure the accuracy of the data?
- What other data sources can be combined to make the data more reliable?
- Can an inpatient event be attributed to a specific cause? How? What confirming data are needed? Can manual review be avoided?
- Are there cases that do not present to the facility?
- How will you capture inpatient events that occur in health systems not participating in the trial?
- Are there effect modifiers (such as age, socioeconomic status, ethnicity, type of medical practice, comorbidity, concomitant medication, and treatment adherence) that will change the treatment effect? (Welsing et al. 2017)
After some initial decisions are made, an investigator will need to consider how to collect the data with as little burden and disruption to the health care system’s workflow as possible (Larson et al. 2015).
Key questions to be considered include:
- Will the act of measuring change clinical practice or the generalizability of the study’s results?
- Do different local practices and workflow result in different interpretations of the same data? How might data availability and interpretation differ by facility, unit or provider?
- Are the documentation procedures consistent across facilities, units, and providers?
- Are there monetary or time costs associated?
- If, after considering these questions, it does not seem practicable to measure the selected outcome, an investigator may need to reconsider what to measure.
Case Studies
In this section, we use case studies to illustrate some of the challenges faced in pragmatic trials that use inpatient events as endpoints.
1. ICD-Pieces: Understanding how re-imbursements affect outcomes measurement
As described earlier in this section, Medicare defines stays in the hospital as either inpatient, outpatient, emergency department, or observation stays.
Medicare Part A pays for inpatient care at a hospital. In 2025, before Medicare starts covering the first 60 days of an inpatient hospital stay (in a single benefit period), a beneficiary must pay a deductible of $1,676.
Medicare Part B pays for outpatient care or care received under observation status. Under Medicare Part B, after paying the annual deductible of $257 (in 2025), a typical patient pays 20% of the Medicare-approved amount for most outpatient and observation-status services under original Medicare (Medicare Interactive 2026).
To reduce costs, in 2016, Medicare updated the rule regarding when inpatient admissions are appropriate for payment under Medicare Part A:
Two-Midnight rule: “Under the Two-Midnight Rule, inpatient admissions are generally payable under Medicare Part A when the admitting physician reasonably expects the patient to require hospital care spanning at least two midnights, and the medical record supports that expectation. A case-by-case exception may also apply for shorter stays when the clinical circumstances warrant inpatient care (Centers for Medicare & Medicaid Services, 2023).” Note that Medicare considers observation care an outpatient service, even if that patient stays overnight in the hospital. In addition, Medicare pays for nursing home care only if a patient spends at least three consecutive days in the hospital as an admitted patient, and observation care doesn’t count on that clock (Medicare Interactive 2019).
This Two Midnight rule differs from definitions currently used by clinical researchers and should be considered when investigators are designing a pragmatic clinical trial. For example, the Improving Chronic Disease Management with Pieces (ICD-Pieces) trial was designed to determine if clinical decision support tools (PIECES) used by practice facilitators can improve the outcomes of patients with chronic kidney disease (CKD), diabetes, and hypertension. The primary outcome is the one-year hospitalization rate (unanticipated hospitalization based on the hospital wide readmission standard from CMS; see definition below). Secondary outcomes include ED visits, readmissions, CV events and death. About two years into the trial, the two-midnight rule was implemented. The original planned primary outcome was unanticipated hospitalization based on the hospital-wide re-admission standard from CMS. Early on in the planning year (the UH2 phase), because of implementation of a new criteria for a “2 midnight rule,” some hospital systems were shifting admissions into observation status (Vazquez and Oliver 2019).
Because of this change in criteria, the investigators defined an un-anticipated hospitalization as both an observational stay and the unplanned hospitalization stay (Vazquez and Oliver 2019). The ICD-pieces team was able to modify their definition of hospitalization as an endpoint to make it more accurately reflect actual care (Vazquez and Oliver 2019).
CMS defines a hospital re-admission as occurring for Medicare beneficiaries who have a readmission for any cause, except for certain planned readmissions, within 30 days from the date of discharge after an eligible index admission. If a beneficiary has more than one unplanned admission (for any reason) within 30 days after discharge from the index admission, only one is counted as a readmission (Centers for Medicare and Medicaid Services 2016).
CMS guidance for reporting outcomes has gone from just hospitalization and re-admission to a new standard: excess days in acute care. In response, health systems have kept patients in observation care or emergency departments. This is important because Medicare’s hospitalization definition may differ from that used in clinical practice.
- Part A (hospital insurance) pays for care received at the hospital by an inpatient.
- Part B (medical insurance) pays for care received at the hospital by an outpatient under observation status.
- Part C (Medicare Advantage) includes both Parts A and B but is provided by private insurance companies.
2. TRANSFORM-HF
The ToRsemide compArisoN with furoSemide FOR Management of Heart Failure (TRANSFORM-HF) trial (ClinicalTrials.gov Identifier: NCT03296813) was an individually randomized PCT for patients hospitalized for new or worsening heart failure (Greene et al. 2021). Prior to hospital discharge, patients received a prescription for one of two commonly prescribed oral diuretics (torsemide or furosemide). All-cause mortality (see Using Death as an Endpoint) and re-hospitalization over 12 months were endpoints.
A re-hospitalization event was defined as either: (1) “an admission to an inpatient unit or a visit to an emergency department that resulted in at least a 24-hour stay (or a change in calendar date if the time of admission/discharge was not available) or (2) an inpatient admission reported by the patient/proxy with an admission date after discharge from the index hospitalization. This definition excluded observational stays of less than 24 hours. The type and reason for the admission were not factored into the TRANSFORM-HF statistical analysis plan's definition of an event.
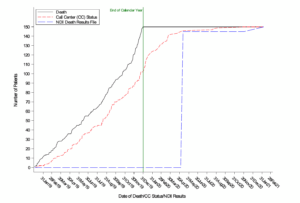
Re-hospitalization events could be triggered via telephone interviews at 1, 6, and 12 months with the patient or proxy, through official medical records obtained when neither the patient nor any of their proxies could be reached, or through discharge summaries from a 12-month medical record query that screened for hospitalizations 12 months after randomization at the patient’s hospital/site (Figure; TRANSFORM HF Statistical Analysis Plan). If an event was triggered, then the event was recorded in the database along with the date and time of admission and discharge.
Figure. TRANSFORM-HF Hospitalization Events

It is worth noting the process for identifying hospitalizations that occurred in health systems other than the systems participating in the trial: if at one of the follow-up calls a patient or proxy indicated that a hospitalization occurred, the study staff requested a discharge summary from sites that were not participating in the trial.
3. Case Example: ADAPTABLE
The Aspirin Dosing: A Patient-Centric Trial Assessing Benefits and Long-term Effectiveness (ADAPTABLE) study was a large, pragmatic trial designed to determine the optimal maintenance aspirin dose for patients with coronary artery disease. According to the protocol, the primary endpoint of this study was the composite rate of all-cause mortality, hospitalization for nonfatal MI, or hospitalization for nonfatal stroke (National Patient-Centered Clinical Research Network (PCORnet) 2015). The sites participating in ADAPTABLE were part of PCORnet, and have agreed to format their data according to a common data model (CDM). Algorithms were validated for extracting endpoint data from the EHR through the CDM, from Medicare claims data, and from select private health plan data (Marquis-Gravel et al. 2021). For more on longitudinal data linkage in ADAPTABLE, see this Case Study. ADAPTABLE enrolled around 15,072 participants (Jones et al. 2021) referred by their physician, and enrolled through an online portal. As a part of this process, they are asked to report any hospitalizations through the portal.
The reconciliation of these patient-reported hospitalizations is shown in the figure below.

From the ADAPTABLE Protocol. Used with permission.
The ADAPTABLE investigators researched patient-reported health data and meta-data standards for patient-reported outcome (PRO) data. They found no useful standardized concepts for hospitalization for nonfatal myocardial infarction (MI) or stroke and state “We do not believe there is much to be gained for the purposes of this study by combining existing atomic concepts to represent these more complex items” (Yang and Tenenbaum 2018).
SECTIONS
REFERENCES
Allen LA, Hernandez AF, O'Connor CM, Felker GM. End points for clinical trials in acute heart failure syndromes. J Am Coll Cardiol. 2009;53(24):2248-2258. doi:10.1016/j.jacc.2008.12.079. PMID: 19520247.
Califf RM, Sugarman J. 2015. Exploring the ethical and regulatory issues in pragmatic clinical trials. Clin Trials. 12:436-441. doi:10.1177/1740774515598334. PMID: 26374676.
Centers for Medicare and Medicaid Services. 2015. Fact sheet: Two-Midnight Rule. https://www.cms.gov/newsroom/fact-sheets/fact-sheet-two-midnight-rule-0
Centers for Medicare and Medicaid Services. 2016. Hospital-Wide All-Cause Unplanned Readmission Measure (NQF #1789). https://innovation.cms.gov/files/fact-sheet/bpciadvanced-fs-nqf1789.pdf
Centers for Medicare & Medicaid Services. 2023. Medicare program: Hospital inpatient prospective payment systems for acute care hospitals and the long-term care hospital prospective payment system and policy changes and fiscal year 2024 rates; Quality reporting and Medicare and Medicaid promoting interoperability programs requirements for eligible hospitals and critical access hospitals; Costs reporting and provider enrollment policies; and physician-owned hospitals. Federal Register, 88(174), 58640–59254. https://www.federalregister.gov/d/2023-18695
Chin CT, Wang TY, Anstrom KJ, et al. 2011. Treatment with adenosine diphosphate receptor inhibitors-longitudinal assessment of treatment patterns and events after acute coronary syndrome (TRANSLATE-ACS) study design: expanding the paradigm of longitudinal observational research. Am Heart J. 162:844–851. doi:10.1016/j.ahj.2011.08.021. PMID: 22093200.
Greene SJ, Velazquez EJ, Anstrom KJ, et al. 2021. Pragmatic design of randomized clinical trials for heart failure. JACC: Heart Failure. 9:325–335. doi:10.1016/j.jchf.2021.01.013. PMID: 33714745.
Hicks KA, Mahaffey KW, Mehran R, et al. 2018. 2017 Cardiovascular and stroke endpoint definitions for clinical trials. Circ. 137:961-972. doi:10.1161/CIRCULATIONAHA.117.033502. PMID: 29483172.
Jones WS, Mulder H, Wruck LM, et al. 2021. Comparative effectiveness of aspirin dosing in cardiovascular disease. N Engl J Med. 384:1981-1990. doi:10.1056/NEJMoa2102137. PMID: 33999548.
Larson EB, Tachibana C, Thompson E, et al. 2015 Jul. Trials without tribulations: minimizing the burden of pragmatic research on healthcare systems. Healthcare. doi:10.1016/j.hjdsi.2015.07.005. PMID: 27637816.
Medicare Interactive. Medicare Interactive. https://www.medicareinteractive.org/. Accessed March 2, 2026.
Marquis-Gravel G, Hammill BG, Mulder H, et al. 2021. Validation of cardiovascular end points ascertainment leveraging multisource electronic health records harmonized into a common data model in the ADAPTABLE randomized clinical trial. Circ Cardiovasc Qual Outcomes. 14:e008190. doi:10.1161/CIRCOUTCOMES.121.008190. PMID: 34886680.
National Patient-Centered Clinical Research Network (PCORnet). 2015. Aspirin Dosing: A Patient-Centric Trial Assessing Benefits and Long-term Effectiveness (ADAPTABLE) Study Protocol.
Perkins DO, Wyatt RJ, Bartko JJ. 2000. Penny-wise and pound-foolish: the impact of measurement error on sample size requirements in clinical trials. Biol Psychiatry. 47:762–766.
U.S. Department of Health and Human Services. 2018. Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics Guidance for Industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics
US Food and Drug Administration. 2018. Framework for FDA’s real-world evidence program. https://www.fda.gov/media/120060/download.
Vazquez MA, Oliver G. 2019. ICD-Pieces: Lessons Learned in an Ongoing Trial. https://dcricollab.dcri.duke.edu/sites/NIHKR/KR/GR-Slides-03-29-19.pdf
Welsing PM, Oude Rengerink K, Collier S, et al. 2017. Series: pragmatic trials and real world evidence: Paper 6. Outcome measures in the real world. J Clin Epidemiol. 90:99-107. doi:10.1016/j.jclinepi.2016.12.022. PMID: 28502810.
Yang Z, Tenenbaum J. 2018. ADAPTABLE Supplement Report: Patient-Reported Health Data and Metadata Standards in the ADAPTABLE Study. https://dcricollab.dcri.duke.edu/sites/NIHKR/KR/ADAPTABLE%20Supplement%20Patient-Reported%20Health%20Data%20Standards-2018-07-19.pdf