The following real-life case study elaborates on each of the recruitment considerations outlined in the previous section. Using a NIH Collaboratory Trial from the NIH Collaboratory as a case study, recruitment considerations in the Improving Chronic Disease Management with Pieces (ICD-Pieces) trial are presented to help illustrate how these questions are being addressed. ICD-Pieces is a cluster randomized clinical trial in patients who have the three coexistent chronic conditions of chronic kidney disease (CKD), Type 2 diabetes, and hypertension. The study tests the hypothesis that practice facilitators can use information technology to identify patients with the triad of CKD, Type 2 diabetes, and hypertension from the electronic health record (EHR) and can assist primary practitioners to deliver evidence-based interventions to improve outcomes compared to usual care.
Recruitment Targets
- Patients 18 to 85 years of age with the three coexisting conditions identified above who receive care from primary care physicians (PCPs) in a participating healthcare system
- Individual practices that are randomized into the intervention group or usual care group within each healthcare system
- Four health systems: Parkland Health, Texas Health Resources, ProHealth Physicians of Connecticut, and the VA North Texas Health Care System
Recruitment Methods
Information Technology Platform
The trial’s recruitment method is based on an electronic patient identification algorithm within Pieces™, a software platform created by the Parkland Center for Clinical Innovation (https://pccinnovation.org). First, the study team searches for prospective eligible patients by screening data extracts from participating clinics’ and providers’ patients. Then, using the electronic tool to create a digitally curated patient registry, eligible patients who have the three medical conditions (i.e., CKD, hypertension, diabetes) are identified at each site. Eligibility criteria include laboratory values, prescription medications, and diagnosis codes for hypertension and diabetes, in addition to laboratory results that establish evidence of chronic kidney disease, including a decrease in estimated glomerular filtration rate (eGFR) and/or proteinuria.
Design and Randomization
The study uses a prospective cluster-randomized design, stratified by the 4 participating healthcare systems, to assign patients to the intervention or control arms. The unit of randomization is the primary care practice. In two of the healthcare systems, clinical practices are defined by an individual PCP caring for a unique panel of patients with a designated clinical team that consists of one RN and one medical assistant, and which does not overlap with the practices of other providers. In the other two systems, clinical practices are defined as a group of patients cared for by providers sharing personnel and workflows at the same practice location and randomized as a single unit.
After stratifying by healthcare system, primary care practices are randomly allocated to either the intervention group or the control (usual care) group using a randomized permutation block within each stratum. Then, based on the assignment of the practice, eligible patients are assigned to either the intervention or control group. Patients in the active intervention arm receive a collaborative model of care facilitated by the Pieces platform and the local Practice Facilitator (described below); patients in the control arm receive treatment as usual.
The primary outcome is the 12-month all-cause hospitalization rate for patients with a triad of CKD, Type 2 diabetes, and hypertension.
The number of patients to be studied totals 10,659, distributed as shown in the following table.
ICD-Pieces Trial Planned Enrollment
| Healthcare system |
Number of practices |
Number of patients to be enrolled |
| ProHealth Connecticut |
57 |
3085 |
| Texas Health Resources |
5 |
3501 |
| Parkland Health and Hospital System |
42 |
3265 |
| VA North Texas Health Care System |
9 |
808 |
Patient Inclusion Criteria
In order to participate in the study, patients must be 18 to 85 years of age and have coexisting CKD, hypertension, and diabetes per the following inclusion criteria:
- CKD (present ≥3 months apart): Two or more eGFRs <60 ml/min or two or more positive tests for albuminuria and/or proteinuria. Albuminuria/proteinuria can be defined by quantitative criteria with albumin/creatinine ratio >30mg/g, urine protein creatinine ratio >200mg/g, or positive dipstick with protein detection (adjusted for urinary concentration/specific gravity).
- Hypertension: Systolic blood pressure >140 mmHg recorded on two separate occasions at least 1 week apart; diastolic blood pressure >90 mmHg on two separate occasions at least 1 week apart; use of antihypertensive agents other than thiazide diuretics, or hypertension included in the problem list within a patient’s medical record. (Thiazide diuretics are excluded because they are commonly prescribed for other conditions such as hypocalcemia, and documented high systolic or diastolic blood pressure in the patient’s chart or hypertension on the problem list in the EHR are more sensitive indicators of patients with hypertension.)
- Diabetes: Random blood glucose >200mg/dL; hemoglobin A1C >6.5%; use of hypoglycemic agents other than metformin, or type 2 diabetes included in the problem list within a patient’s medical record. (Metformin is excluded because it is commonly prescribed for other conditions such as polycystic ovary disease, and documented abnormal lab results in the patient’s chart, or type 2 diabetes on the problem list are more sensitive indicators of patients with type 2 diabetes.)
The study team created a registry of eligible patients and updates the database on schedule for each site. The software tool (Pieces) identifies patients meeting study criteria from the EHR and sends data files of both candidate and confirmed patients on a weekly basis to the ICD-Pieces registry created within the EHR. The registry lists eligible patients and their information such as demographics, lab results and dates, diagnosis on problem list, medication list, and date of next PCP visit. Candidate patients are those confirmed to have type 2 diabetes and hypertension but need a laboratory result within 12 months of their visit to confirm that they have CKD. Confirmed patients are those who have received laboratory confirmation of CKD within the past 12 months of their visit, in addition to having diagnoses of hypertension and type 2 diabetes.
Practice Facilitators
In coordination with the study’s local Practice Facilitator, participating healthcare systems have flexibility in the method of notifying physicians about upcoming clinic appointments for eligible patients. The role of Practice Facilitator is essential in supporting the study’s patient enrollment, patient education, and monitoring to ensure that patients receive the necessary intervention within the course of normal clinic operations and that they progress to care targets. At two of the participating healthcare systems, the facilitator is an RN. At the other two systems, the facilitator is a pharmacist with support from research assistants. The facilitator uses the study registry to identify both candidate and confirmed patients who have an office visit scheduled within the following 1 to 2 weeks. For previsit planning, the facilitator notifies the office staff of candidate patients who need labs completed before their office visit. The office staff then contacts patients who need a lab test to have it done in advance of their clinic visit.
For confirmed patients, providers are notified in any of three ways based on available IT resources:
- An embedded decision support/Best Practices Alert during the patient visit
- An individual study enrollment notification to the provider by pharmacist notes within the patient chart
- Group messaging to providers from the facilitator with a list of all the providers' confirmed eligible patients who have clinic appointments scheduled for that week
Primary Care Provider Participation
Recruitment of patients into the ICD-Pieces study is dependent on the participation of providers. In all 4 systems, provider recruitment began with soliciting and obtaining agreement from the executive leadership of each healthcare system. Involvement of and support by executive leadership was critical to promote acceptance by providers. Criteria for selecting providers to participate in the study include:
- Connection of the clinic or practice to the healthcare system’s main EHR platform (e.g., Epic, Allscripts) in order to obtain patient data
- Organization and clinic readiness as determined by leadership of each healthcare system (e.g., considering factors such as inadequate staffing, competing quality initiative programs, clinic reorganization and restructuring)
The four participating healthcare systems designed different methods for recruiting providers into the study according to their system’s operations. Providers are informed about the ICD-Pieces study, including their expected roles, at their scheduled medical or clinical meetings. During these sessions, they can ask questions and seek clarification from the study team. Providers can decline from enrolling their patients. The focus is on minimizing any burden of participating in the clinical trial.
The unit of randomization for the trial is at the primary care practice level. Practices that were randomized into the intervention group were onboarded at different times to begin patient recruitment. The provider has the final say in decision making about patient enrollment. At 2 of the healthcare systems, the providers receive notification during clinic visits of their confirmed eligible patients, and they can decline enrollment if they think their patient is not suitable for the study. At the other two healthcare systems, providers are notified by a pharmacist note appended to the patient’s chart, and then the provider can choose to implement or decline the recommendations. After providers agree to enroll their patients in the study, providers will activate the study protocol by signing the study orders for patient care. At some of the clinic sites, the clinic staff may pin the orders to patient charts as reminder for providers to sign the orders.
Patient recruitment has been affected by provider turnover due to retirement, relocation, or ceasing their affiliation with the healthcare systems. The study has contingency plans in place for provider replacement.
Informed Consent and Opt Out
A waiver of informed consent was approved by the three Institutional Review Boards (IRBs) overseeing this study. Information about the study is available to patients in the participating practices. To respect potential patient privacy concerns, patients are offered the opportunity, using public notification of available opt-out mechanisms, to opt out of their data being used in the study. The office staff hands out educational materials and a patient information sheet that describes the study and the research team to eligible patients, plus a contact phone number to call if they decide to opt out of the study.
The following figure shows a diagram of a sample workflow for a healthcare system participating in the ICD-Pieces trial.
Workflow for Initial Patient Visit
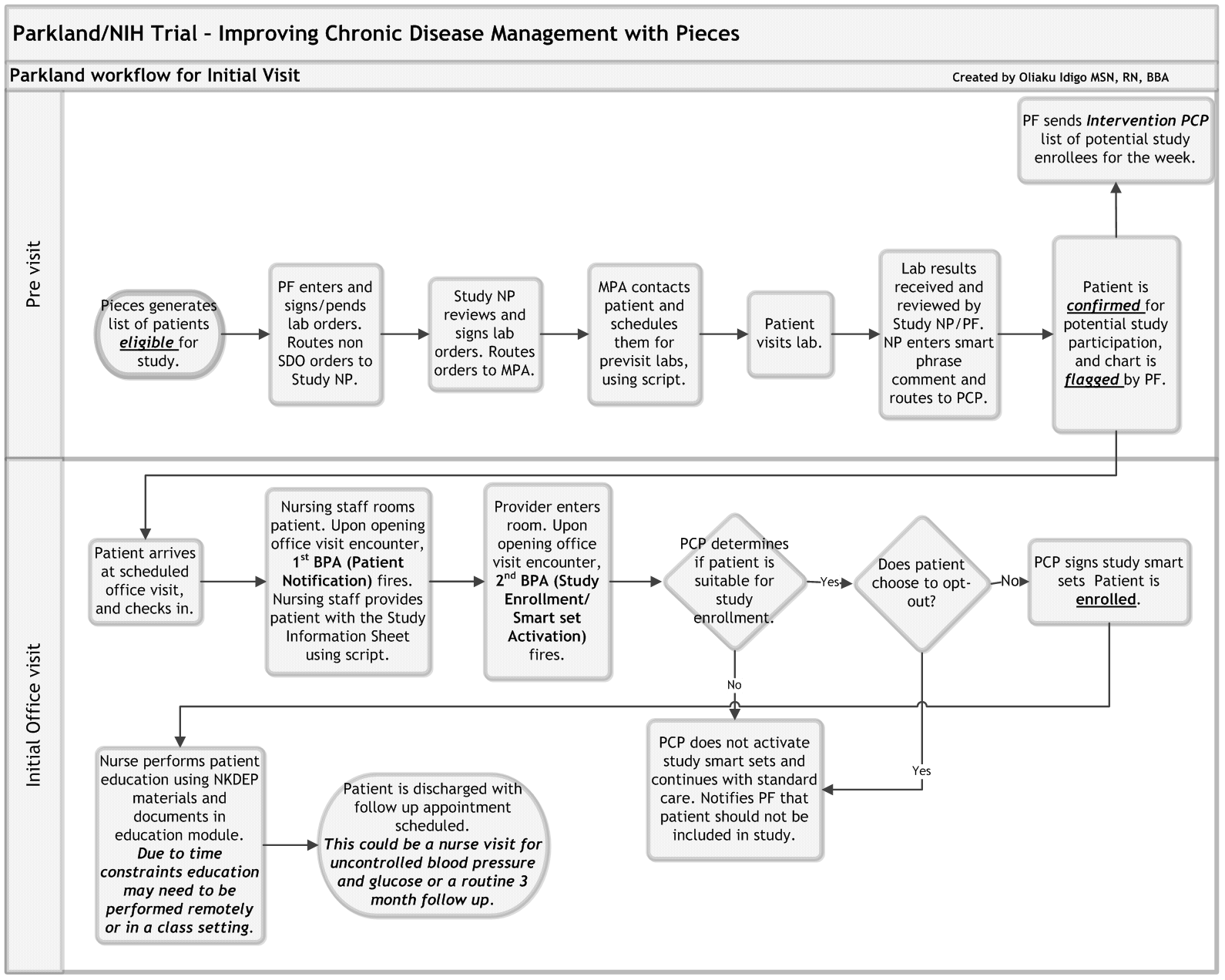
* Diagram created by Oliaku Idigo, MSN, RN, BBA, Parkland Health and Hospital System.
Abbreviations: BPA=Best Practice Alert; MPA=Medical Practice Assistant; NP=Nurse Practitioner; PCP=Primary Care Physician; PF=Practice Facilitator
Recruitment Materials
- Pieces™ database
- ICD-Pieces study registry
- Epic® Reporting Workbench
- Best Practice Alerts
- Study protocols including the CKD diabetes management protocol and the CKD hypertension management protocol
- Order sets/smart sets created within the EHR
- Pharmacist notes in the EHR
Enrolling Participants
The Practice Facilitator contacts the office manager to schedule a face-to-face onboarding meeting with providers and clinic staff at the time of their regular office meeting to discuss the study, and the role providers and staff are expected to play in patient enrollment. The meeting also is essential to obtain input on how to fit study activities into their clinical workflow. This is followed by ongoing contact via e-mail, telephone, or in-basket messaging (a secure, closed messaging system within the EHR used for sending and receiving messages about patient care and billing needs).
Patient enrollment in the intervention group occurs during the clinic visit when the PCP activates the study protocol and signs the study orders using the Best Practice Alert that appears during the office visit or acknowledges the pharmacist notes. Order sets (also known as smart sets)—a group of related orders that apply to a prespecified diagnosis or particular period of time—are activated and delegated by PCPs as standing orders, such as blood pressure monitoring, patient education, and medication titration. Patients in the usual care group have to meet study inclusion criteria and be seen in clinic.
Although eligibility for enrollment is determined before the patient's office visit, PCPs have the final say as to whether their patients are suitable for the study and PCP exclusions are tracked by the study team. We recognize that we may not be able to enumerate all patients’ conditions that need to be excluded (e.g., terminal cancer, cirrhosis, or other terminal conditions). After order set activation, the office staff and Practice Facilitator provide patient education and initiate follow-up procedures leading up to the next scheduled office visit.
Use of the Electronic Health Record System
The study team created a study registry as well as a best practice alert, smart set, and study protocol in the Epic EHR at 2 of the participating healthcare systems, Parkland and Texas Health Resources. Pharmacist note templates were created at the VA and ProHealth systems. Regular automated updates are entered into the study database from the EHR, which enables the continuous monitoring and assessment of enrolled patients’ progress toward the study goals in the intervention group. The study team plans to use the EHR to retrieve data for final outcome assessment.
The primary outcome of ICD-Pieces is all-cause hospitalizations for patients with a triad of CKD, diabetes, and hypertension. Secondary outcomes include 30-day all-cause readmissions (for those patients who have an index hospitalization), emergency room visits, cardiovascular events, dialysis and deaths.
Potential Barriers and Challenges
ICD-Pieces Recruitment Design Modification
The randomization for ICD-Pieces was initially planned to be based on clinics of the four healthcare systems involved in the study. However, it was discovered that the patient panels in clinics varied in size among systems. This introduced an element of heterogeneity among clusters being randomized and negatively affected the intracluster correlation coefficient (ICC) and effect sample size. Even with a large number of eligible patients, it could be difficult to detect a difference between the intervention and usual care groups.
For example, at Parkland Health and Hospital System, the early plans for panel size for clinics ranged between 300 to more than 1000 patients per cluster due to the larger number of providers employed at some of the clinics. In comparison, the patient panel size for Texas Health Resources clinics was mostly under 100. The organization of clusters at Parkland was redefined to practices based on patient panels managed by unique teams of one provider and staff. As a consequence, it was possible to achieve more homogeneous clusters and an effect study sample size with higher power to detect a difference. At ProHealth Physicians of Connecticut, administrative adjustments in division of clinic regions led to an adjustment in cluster sizes from 13 regional consolidated clinics to 50 distinct practices or clusters, resulting in clusters similar in size to practices in the other systems participating in the trial.
A possible concern with cluster randomization by practices is cross-contamination between intervention and usual care groups (i.e., the clinical practices in this case). In ICD-Pieces, this risk is mitigated by the fact that each individual practice cares for a unique group of patients with a permanently designated team of one RN and one medical assistant, and does not overlap with other practices. The ICD-Pieces team generates patient lists/registries for each individual practice, allowing Practice Facilitators to direct interventions to the intervention group and avoid generalized alerts/activation of protocols and cross-contamination to the control group.
Providers’ Lack of Understanding of Waiver of Consent Concept
The study received a waiver of informed consent from the three IRBs overseeing the study. However, patients are able to opt out of their data being used in the study, and providers can opt out patients based on their clinical judgment. Some providers initially struggled with the concept of enrolling patients into the study without written informed consent. A few patients declined to participate when their provider attempted to explain the study to them. It took some effort to engage a few providers before they understood the appropriate study workflow, which begins with the office staff providing an information sheet to each eligible patient before he or she enters the examination room to see his or her provider. The information sheet describes the study, its purpose, the study team, and how patients can opt out of the study if they so choose. The provider’s role is to provide the recommended evidence-based care to his or her patients and then authorize the Practice Facilitator to follow up with patients based on established protocols. After the providers understood the workflow, enrollment of patients began to run smoothly.
Patients Not Appearing for Clinic Appointments
For patients who are “no shows” at their clinic visits, the office staff calls to reschedule the appointment. The proportion of eligible patients missing appointments has varied among healthcare systems.
Shortage of Clinic Staff
Sometimes a staff shortage during busy periods in smaller clinics has the effect of interrupting patient enrollment. The study team endeavors to catch the patients on their next PCP visit.
Data Transmission Issues
Changes in the Pieces data integration system and a turnover of technical staff have the effect of slowing down enrollment. The data integration system is an IT platform for receiving, joining, combining, and analyzing data on a cloud-based server that was migrated from one cloud provider to another. During the trial, it became necessary to change the data transmission pathway and issue new credentials. Another IT issue involved lab data that were not being updated in the study’s registry; consequently, some eligible patients were not identified when they came to their clinic visit. The study team was able to resolve these IT issues and get enrollment back on track.
Providers’ Resistance
Some providers were initially resistant because of the perception that the study adds an inordinate level of burden to their regular workflow. The study team worked with them to better incorporate study activities into the clinic workflow to minimize the burden on providers and staff. The Practice Facilitators took on more responsibilities, such as medication titration and ordering labs as authorized by the provider. In one health system, some providers have become advocates for the study because, after being part of it, they realized it was reducing their burden while helping them meet their performance metrics.
There was also a question about payment to physicians to conduct research. It was necessary to explain that the funds for the ICD-Pieces trial are strictly intended for research and not for reimbursing regular patient care. Enrollment of patients through activation of order sets requires very minimal extra time from PCPs.
Last, the issue of loss of “locus of control” was recognized and respected in all interactions with providers. Providers are accustomed to making decisions about the care provided to their patients and they may feel a loss of control because additional care is being suggested for their patients.
Other Resources for Recruitment
Participant recruitment is one of the most critical plans to design and implement in clinical research, whether it be a more traditional or a more pragmatic trial. This ICD-Pieces case study has described several recruitment details within a pragmatic trial, including electronic platforms and clinical data that can augment recruitment, and various factors that might help or hinder it. Still, other pragmatic trials also provide learning opportunities on enrollment, and some of those resources follow in the next section.
DISCLAIMER: The views expressed in this chapter are those of the contributors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.


